Launching a pipeline
You can launch a Nextflow pipeline by simply executing this code:
nextflow run main.nf --help
We should see something like:
nextflow run main.nf --help
Picked up JAVA_TOOL_OPTIONS: -Xmx2576m
N E X T F L O W ~ version 21.10.3
Launching `main.nf` [distracted_avogadro] - revision: e434616b3d
BIOCORE@CRG - N F TESTPIPE
=============================================
reads : /workspace/elixir-workshop-21/data/*.fastq.gz
reference : /workspace/elixir-workshop-21/data/chr19.fasta.gz
output : ./output
This is the Biocore's Nextflow test pipeline
Please define reads, reference and output!
Enjoy!
But is really convenient to store your code in one of the sharing platform that are supported, so let’s try a different thing.
Sharing Nextflow code
Nextflow supports a number of code sharing platforms: BitBucket, GitHub, and GitLab. This feature allows running pipelines by just pointing to an online repository without caring about downloading, installing etc. The default platform is GitHub, so we will use this platform and in particular this repository as an example.
GitHub |
BitBucket |
GitLab |
|---|---|---|
|
|
|
We can launch the pipeline just by typing:
In this way Nextflow will pull the pipeline from the GitHub repository, store it at $HOME/.nextflow/assets and launch it.
nextflow run nextflow-io/elixir-workshop-21 -r master --help
Picked up JAVA_TOOL_OPTIONS: -Xmx2576m
N E X T F L O W ~ version 21.10.3
Pulling nextflow-io/elixir-workshop-21 ...
downloaded from https://github.com/nextflow-io/elixir-workshop-21.git
Launching `nextflow-io/elixir-workshop-21` [prickly_panini] - revision: 12648e4544 [master]
BIOCORE@CRG - N F TESTPIPE
=============================================
reads : /home/gitpod/.nextflow/assets/nextflow-io/elixir-workshop-21/data/*.fastq.gz
reference : /home/gitpod/.nextflow/assets/nextflow-io/elixir-workshop-21/data/chr19.fasta.gz
output : ./output
This is the Biocore's Nextflow test pipeline
Please define reads, reference and output!
Enjoy!
Linux containers
This pipeline needs tools that are stored within linux containers. In particular we will use images for Bowtie, FastQC and multiQC that are retrieved from Biocontainers.
For this we need to tell Nextflow which is the right container engine. We can do this by using the Nextflow parameter -with-docker.
Nextflow supports many more container engines like Singularity, Shifter, Podman and Charliecloud.
Docker |
Singularity |
Shifter |
Podman |
Charliecloud |
|---|---|---|---|---|
|
|
|
|
|


In this example we will use Docker: the default image is stored in DockerHub and is retrieved on the fly.
nextflow run nextflow-io/elixir-workshop-21 -r master -with-docker
N E X T F L O W ~ version 21.04.3
Pulling nextflow-io/elixir-workshop-21 ...
Launching `nextflow-io/elixir-workshop-21` [determined_minsky] - revision: 711f9f806d [master]
BIOCORE@CRG - N F TESTPIPE ~ version 1.0
=============================================
reads : /Users/lcozzuto/.nextflow/assets/nextflow-io/elixir-workshop-21/data/*.fastq.gz
reference : /Users/lcozzuto/.nextflow/assets/nextflow-io/elixir-workshop-21/data/chr19.fasta.gz
output : /Users/lcozzuto/.nextflow/assets/nextflow-io/elixir-workshop-21/output
executor > local (3)
[2f/6cd1ca] process > fastqc (B7_H3K4me1_s_chr19.fastq.gz) [100%] 2 of 2 ✔
[a2/305aae] process > BOWTIE:Index (chr19.fasta.gz) [ 0%] 0 of 1
[79/f30f70] process > BOWTIE:Align (B7_H3K4me1_s_chr19.fastq.gz) [100%] 2 of 2 ✔
[50/088302] process > multiqc [100%] 1 of 1 ✔
/Users/lcozzuto/work/27/f1cc39c1e01c9ee55684b347c492f5/B7_input_s_chr19.fastq.gz.sam
/Users/lcozzuto/work/27/f1cc39c1e01c9ee55684b347c492f5/B7_input_s_chr19.fastq.gz.log
/Users/lcozzuto/work/79/f30f7041abf5baf7c496a0982906c1/B7_H3K4me1_s_chr19.fastq.gz.sam
/Users/lcozzuto/work/79/f30f7041abf5baf7c496a0982906c1/B7_H3K4me1_s_chr19.fastq.gz.log
Done! Open the following report in your browser --> /Users/lcozzuto/.nextflow/assets/nextflow-io/elixir-workshop-21/output/ouptut_multiQC/multiqc_report.html
Completed at: 24-Nov-2021 15:48:35
Duration : 3m 25s
CPU hours : 0.1
Succeeded : 6
This pipeline can be launched also with Singularity just using the Nextflow parameter -with-singularity. Nextflow will retrieve and convert the image(s) for you. The image(s) will be then stored so that next time you don’t need to download anything again.
We can inspect the output in the new output folder generated.
ls -alht output
ls -alht
total 0
drwxr-xr-x 17 lcozzuto staff 544B Nov 24 16:18 ..
drwxr-xr-x 3 lcozzuto staff 96B Nov 24 16:13 ouptut_multiQC
drwxr-xr-x 5 lcozzuto staff 160B Nov 24 16:13 .
drwxr-xr-x 4 lcozzuto staff 128B Nov 24 16:13 ouptut_aln
drwxr-xr-x 6 lcozzuto staff 192B Nov 24 16:11 ouptut_fastqc
Here you can see the report produced by multiQC. You can download the one you just generated by clicking with the righ button on that file.
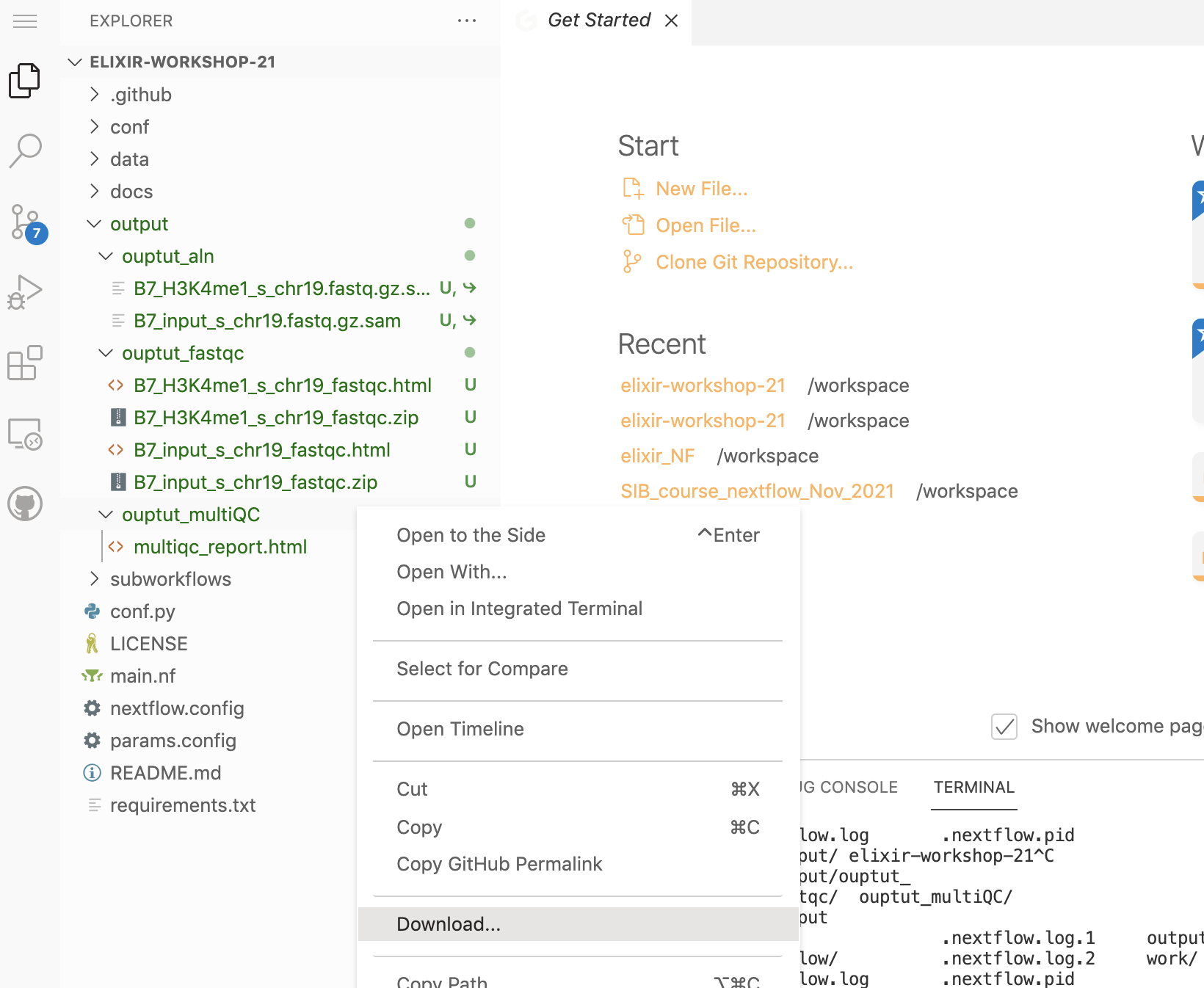
Work folder structure and process isolation
Once executed, we can see that a folder named work is generated. Nextflow stores in this folder the intermediate files generated by each processes. In case you resume a process that folder is “reused” as cache.
At the start of each row, there is an alphanumeric code:
[a2/305aae] process > BOWTIE:Index (chr19.fasta.gz) [ 0%] 0 of 1
This code indicates the path in which the process is “isolated” and where the corresponding temporary files are kept in the work directory.
Note
Nextflow will randomly generate temporary folders so they will be named differently in your execution.
Let’s have a look inside that folder:
cd work/a2/305aaee297250b0c7a455cab35707c/
ls -alht
-rw-r--r-- 1 lcozzuto staff 20M Nov 24 16:12 chr19.fasta.gz.rev.1.ebwt
-rw-r--r-- 1 lcozzuto staff 6.9M Nov 24 16:12 chr19.fasta.gz.rev.2.ebwt
-rw-r--r-- 1 lcozzuto staff 20M Nov 24 16:11 chr19.fasta.gz.1.ebwt
-rw-r--r-- 1 lcozzuto staff 6.9M Nov 24 16:11 chr19.fasta.gz.2.ebwt
-rw-r--r-- 1 lcozzuto staff 53B Nov 24 16:10 chr19.fasta.gz.3.ebwt
-rw-r--r-- 1 lcozzuto staff 14M Nov 24 16:10 chr19.fasta.gz.4.ebwt
lrwxr-xr-x 1 lcozzuto staff 74B Nov 24 16:10 chr19.fasta.gz -> /Users/lcozzuto/.nextflow/assets/nextflow-io/elixir-workshop-21/data/chr19.fasta.gz
You can see the input files staged as links, the output files and some “hidden” files in which we have different information:
.exitcode, contains 0 if everything is ok, another value if there was a problem.
.command.log, contains the log of the command execution. It is often identical to .command.out
.command.out, contains the standard output of the command execution
.command.err, contains the standard error of the command execution
.command.begin, contains what has to be executed before .command.sh
.command.sh, contains the block of code indicated in the process
.command.run, contains the code made by nextflow for the execution of .command.sh, and contains environmental variables, eventual invocations of linux containers etc.
Resuming and changing parameters
We can copy a fastq files in another place and change the file name:
cp $PATH/.nextflow/assets/nextflow-io/elixir-workshop-21/data/*.gz .
mv B7_H3K4me1_s_chr19.fastq.gz test2.fastq.gz
mv B7_input_s_chr19.fastq.gz test1.fastq.gz
Then we can execute again the pipeline feeding the new input files by using the pipeline parameter --reads ""
Note
Nextflow parameters are indicated by one dash (-). Pipeline parameters by two dahses (--)
You can execute again the pipeline by using the Nextflow parameter -resume and send it to background with -bg.
nextflow run nextflow-io/elixir-workshop-21 -with-docker -r master -bg --reads "*.fastq.gz" -resume > log
cat log
N E X T F L O W ~ version 21.10.3
Launching `nextflow-io/elixir-workshop-21` [jolly_visvesvaraya] - revision: 040cd63a79 [master]
NOTE: Your local project version looks outdated - a different revision is available in the remote repository [cf2612db62]
BIOCORE@CRG - N F TESTPIPE ~ version 1.0
=============================================
reads : *.fastq.gz
reference : /Users/lcozzuto/.nextflow/assets/nextflow-io/elixir-workshop-21/data/chr19.fasta.gz
output : ./output
[8b/cfcc4f] Submitted process > fastqc (test1.fastq.gz)
[5b/71ae88] Submitted process > fastqc (test2.fastq.gz)
[6e/1cc3be] Cached process > BOWTIE:Index (chr19.fasta.gz)
[97/2a6a72] Submitted process > BOWTIE:Align (test2.fastq.gz)
[0a/951748] Submitted process > BOWTIE:Align (test1.fastq.gz)
/Users/lcozzuto/work/97/2a6a7245675d7913019aa8983c5e55/test2.fastq.gz.log
/Users/lcozzuto/work/97/2a6a7245675d7913019aa8983c5e55/test2.fastq.gz.sam
/Users/lcozzuto/work/0a/9517481ef43b0e88163ec5f8b4d71f/test1.fastq.gz.log
/Users/lcozzuto/work/0a/9517481ef43b0e88163ec5f8b4d71f/test1.fastq.gz.sam
[f7/1b5746] Submitted process > multiqc
Done! Open the following report in your browser --> ./output/ouptut_multiQC/multiqc_report.html
You can see that the indexing of the genome is cached while the processes that are influenced by the new files are triggered.
Reporting and monitoring
Before going to the code we can have a look to two important features of Nextflow: the ability to produce a comprehensive report and the live monitoring offered by tower.nf web application.
We can go to the tower.nf website

and click on the GitHub authentication.
You can generate your token at https://tower.nf/tokens exporting those environmental variables:
export TOWER_ACCESS_TOKEN=*******YOUR***TOKEN*****HERE*******
Note
You can also store them indefinitely in your .bashrc or .bash_profile file.
We can then launch again the pipeline forcing this time without -resume and check the live reporting on the tower website adding the parameter -with-tower.
nextflow run nextflow-io/elixir-workshop-21 -with-docker -r master -bg --reads "*.fastq.gz" -with-tower > log
tail -f log
N E X T F L O W ~ version 21.10.3
Launching `nextflow-io/elixir-workshop-21` [evil_ekeblad] - revision: 040cd63a79 [master]
NOTE: Your local project version looks outdated - a different revision is available in the remote repository [fb23636633]
Downloading plugin nf-tower@1.3.0
BIOCORE@CRG - N F TESTPIPE ~ version 1.0
=============================================
reads : *.fastq.gz
reference : /Users/lcozzuto/.nextflow/assets/nextflow-io/elixir-workshop-21/data/chr19.fasta.gz
output : ./output
Monitor the execution with Nextflow Tower using this url https://tower.nf/user/lucacozzuto/watch/54kIaLzfwIfiLx
[23/b06dda] Submitted process > fastqc (test1.fastq.gz)
[ee/82bce0] Submitted process > fastqc (test2.fastq.gz)
[27/82af32] Submitted process > BOWTIE:Index (chr19.fasta.gz)
[...]
We can check the appearance of a new pipeline and the content
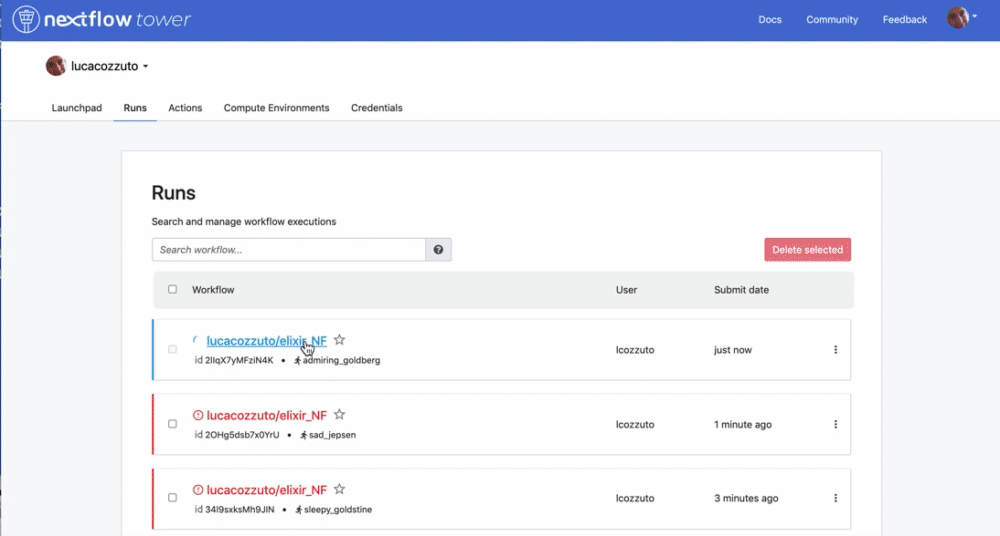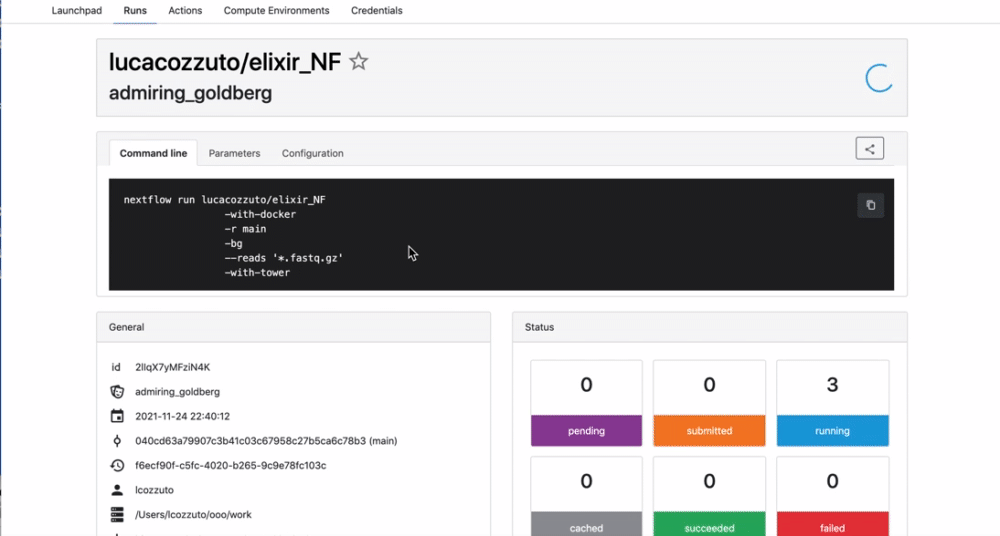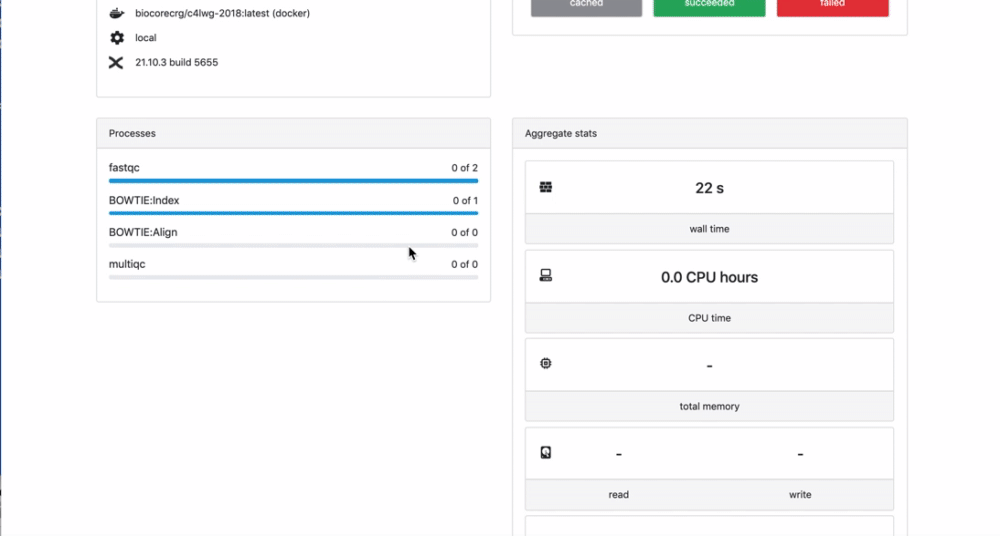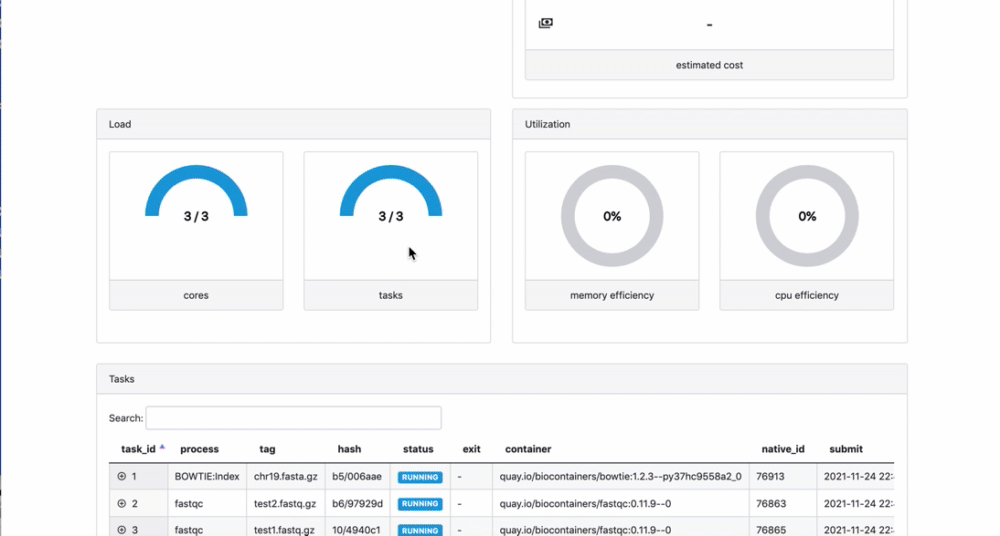
Here you have a real case of a two days run with almost 6 thousands jobs.
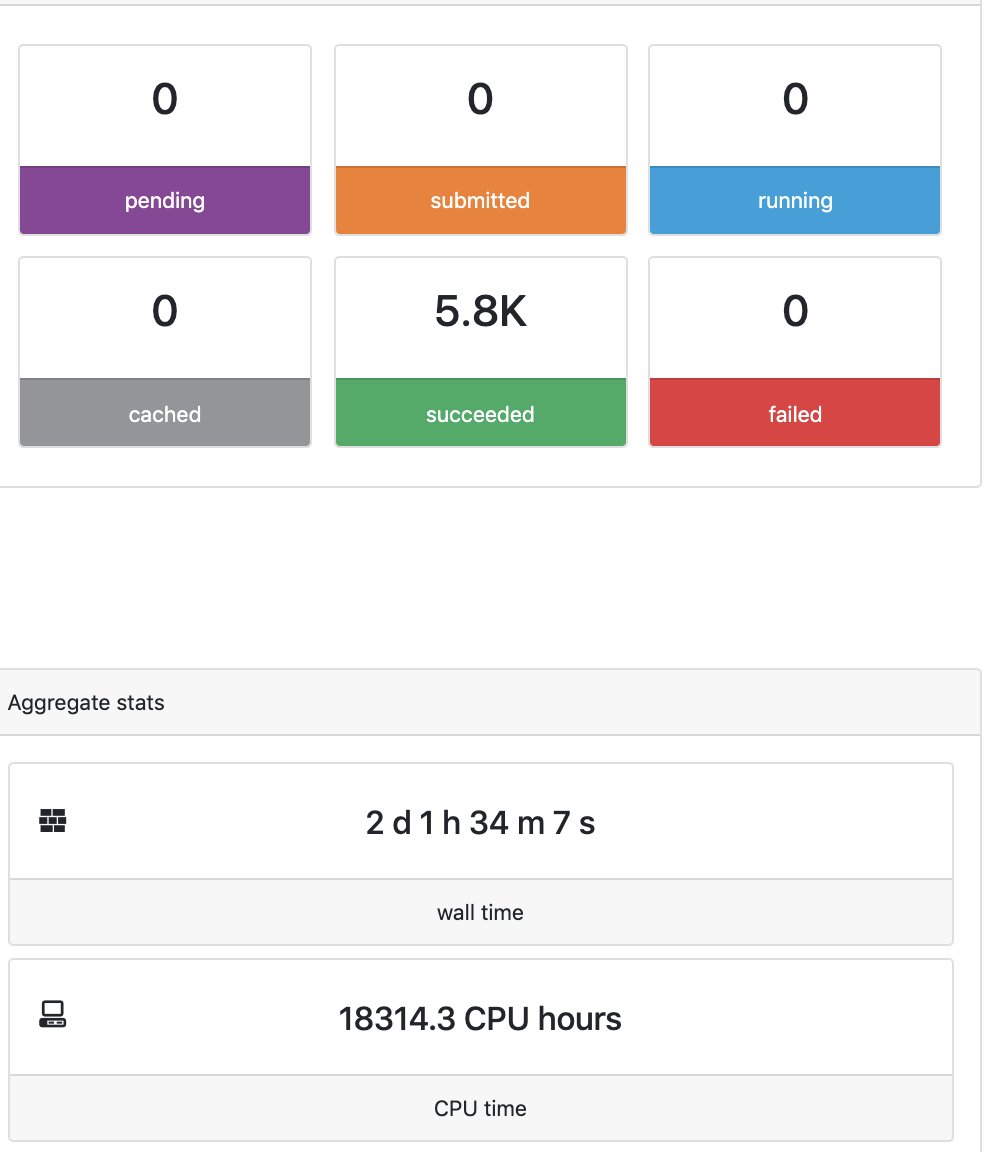
When the pipeline is finished you also get a mail. Adding the parameter -with-report will produce a final html report with all the information that was in the tower.nf website.